End-to-End Processing of a Microbial Metagenome
For this dataset, we’ll be fully processing SRX4071230, a peat metagenome with 14.1M reads. The SRA Run is SRR7151490, which will be important when downloading the data from SRA.
This will include (nearly) all steps and most of the results returned from the command line. Clearly, some outputs can’t be nicely placed here, but are available (as links to files) or in the M8161 project directory.
Everything here uses Singularity. All of the singularity images are located at:
/users/PAS1117/osu9664/eMicro-Apps/
OR
/fs/ess/PAS1573/sif/
You need to either provide full paths to the images/containers, or extend your system’s PATH variable (see Available Environmental Microbiology (“eMicro”) Apps and Tools)
Downloading the data
The first thing we need to do is grab the data from the SRA. You can do this a few ways, either through navigating the NCBI+SRA websites, or directly using their SRA Toolkit.
# Move to project directory
$ cd /fs/ess/PAS1573/week7_processing
# Load modules necessary
$ module load singularity
$ singularity run SRA_Toolkit.sif fasterq-dump –e 4 SRR7151490
spots read : 14,145,898
reads read : 28,291,796
reads written : 28,291,796
In the example above, we used fasterq-dump, which is designated to download two paired end read files in fastq format. We also specified 4 threads (-e 4) so it would run a little faster. There should be two output files: SRR7151490_1.fastq and SRR7151490_2.fastq. fasterq-dump won’t compress the files for you, so you’ll have to do this after the download completes.
Alternatively, you can manually download the data from the NCBI website:
Go to https://www.ncbi.nlm.nih.gov/, and under the search bar, select BioProject and type “PRJNA386568”. It should immediately pull up a single page:
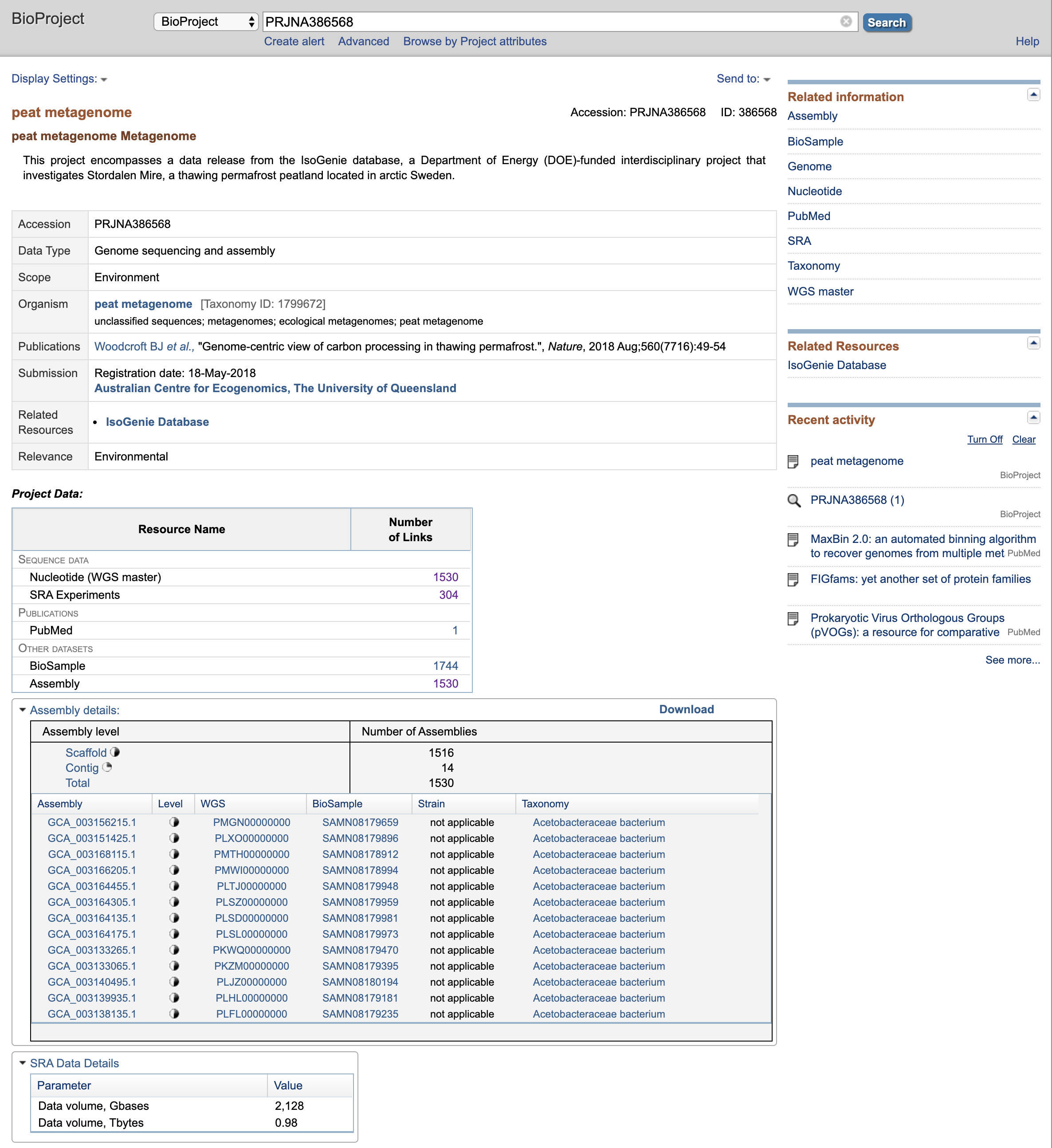
Nagivate to SRA Experiments (should be 304 of them), select. This should pull up a list of… SRA experiments.
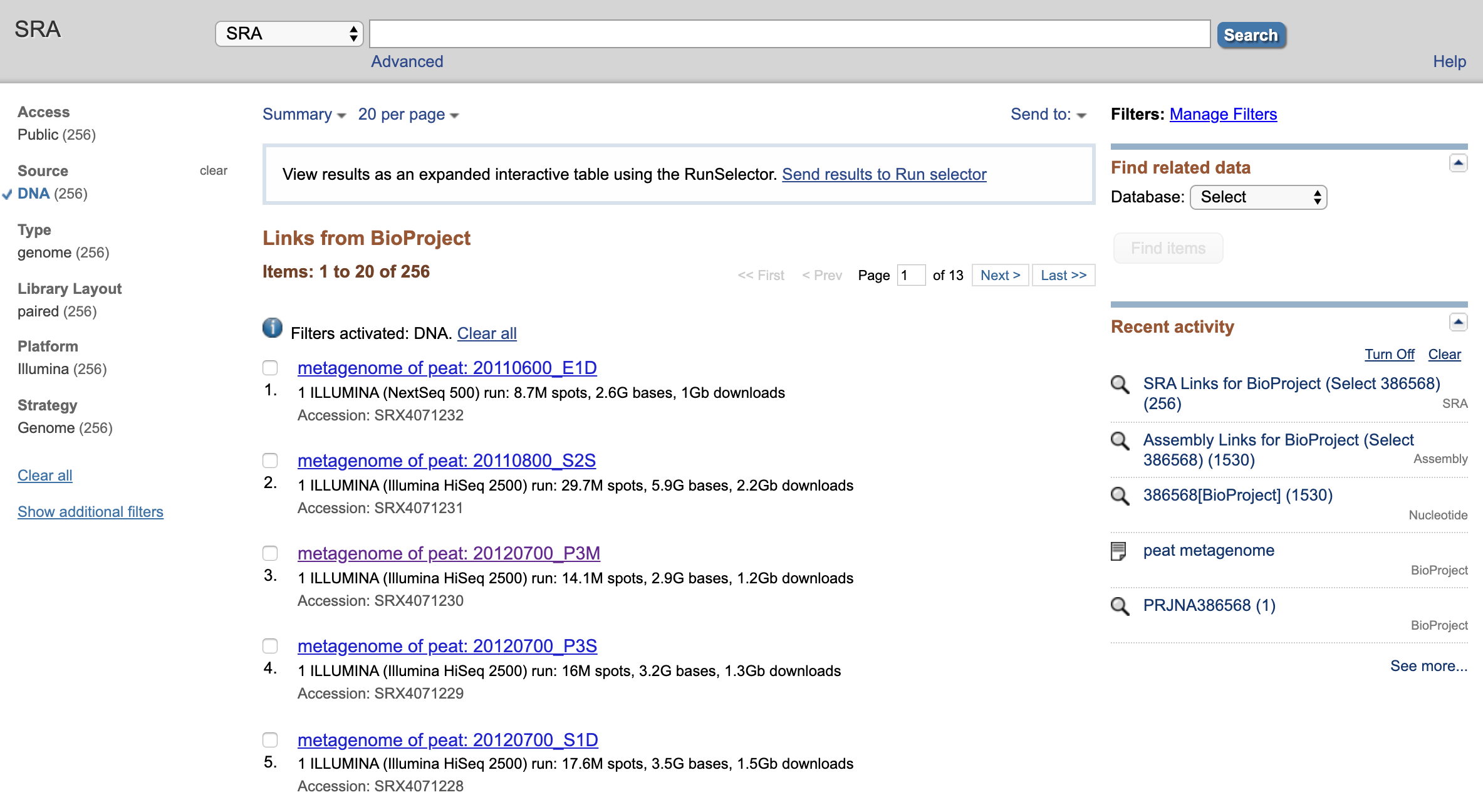
Select (or find) “metagenome of peat: 20120700_P3M”. Select.
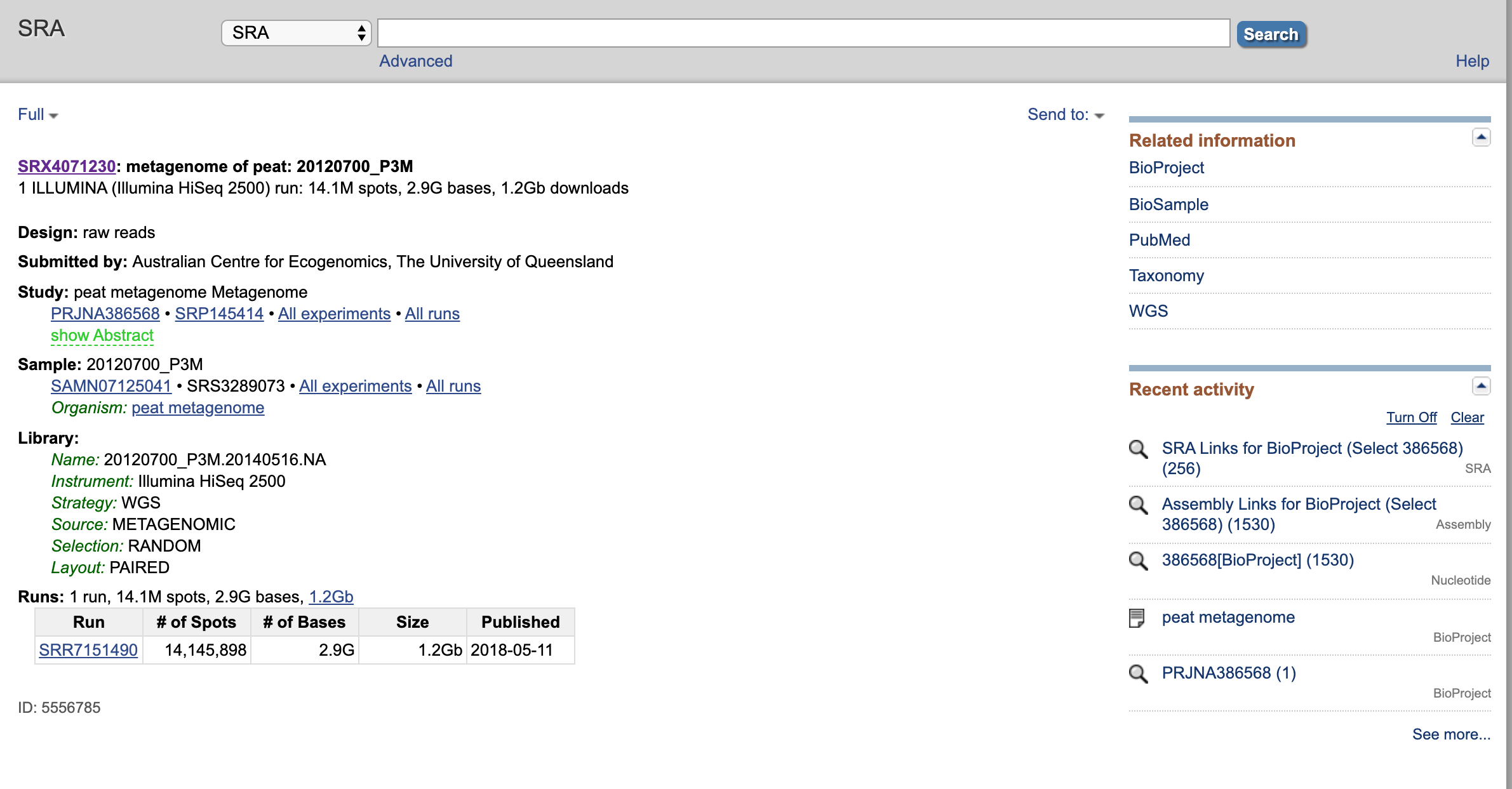
Now we have some info about the SRA experiment. Click on the Run (should start with SRR) towards the bottom of the metadata fields and select the run.
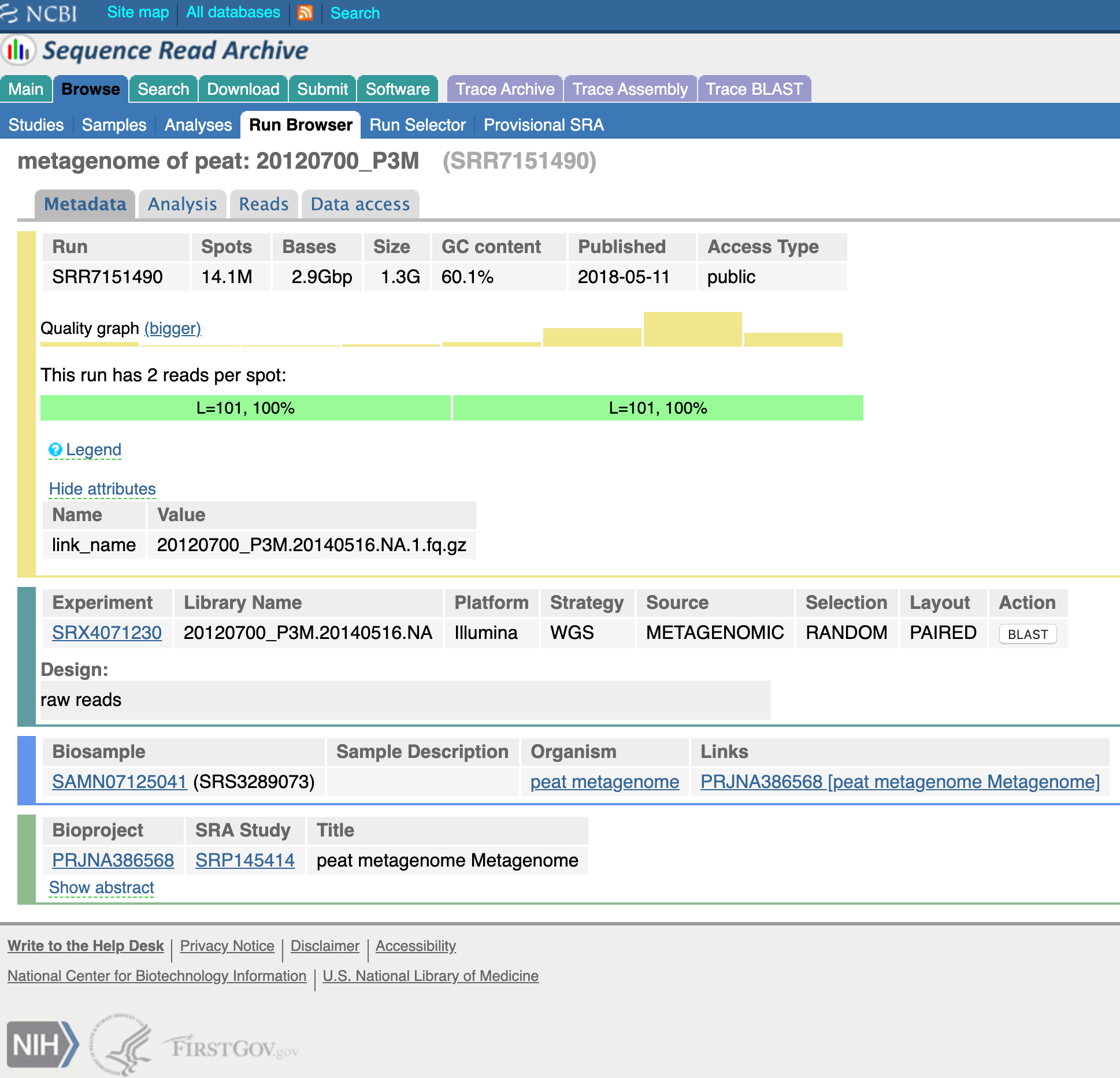
Click on the “Data access” tab and voila, a download will start.
Read Quality Control
We will be using either BBDuk or Trimmomatic to process our input reads. You only need to select one. We’ll be using both for examples, but typically stick with one and use it.
$ singularity run Trimmomatic-0.36.0.img PE SRR7151490_1.fastq SRR7151490_2.fastq SRR7151490_1_t_paired.fastq.gz SRR7151490_1_t_unpaired.fastq.gz SRR7151490_2_t_paired.fastq.gz SRR7151490_2_t_unpaired.fastq.gz ILLUMINACLIP:/Trimmomatic-0.36/adapters/TruSeq3-PE.fa:2:30:10:2 LEADING:3 TRAILING:3 MINLEN:36
TrimmomaticPE: Started with arguments:
SRR7151490_1.fastq SRR7151490_2.fastq SRR7151490_1_t_paired.fastq.gz SRR7151490_1_t_unpaired.fastq.gz SRR7151490_2_t_paired.fastq.gz SRR7151490_2_t_unpaired.fastq.gz ILLUMINACLIP:/Trimmomatic-0.36/adapters/TruSeq3-PE.fa:2:30:10:2 LEADING:3 TRAILING:3 MINLEN:36
Using PrefixPair: 'TACACTCTTTCCCTACACGACGCTCTTCCGATCT' and 'GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT'
ILLUMINACLIP: Using 1 prefix pairs, 0 forward/reverse sequences, 0 forward only sequences, 0 reverse only sequences
Quality encoding detected as phred33
Input Read Pairs: 14145898 Both Surviving: 13356639 (94.42%) Forward Only Surviving: 705239 (4.99%) Reverse Only Surviving: 64641 (0.46%) Dropped: 19379 (0.14%)
TrimmomaticPE: Completed successfully
real 11m0.811s
user 11m43.109s
sys 0m12.327s
For Trimmomatic, the defaults work pretty well. Note the location of the IlluminaClip - it’s already “in” the Singularity file. If you have your own custom primers/adapters, you’ll need to add your sequences or create your own primer and adapter file.
Next, BBDuk. BBDuk is usually done in 2-3 steps, with 1st being an adapter trimming step, and 2nd with the removal of low quality sequences. You can do both steps in a single command, but doing so in two steps allows us to see what was removed during each.
$ singularity run BBTools-38.69.sif bbduk.sh in1=SRR7151490_1.fastq in2=SRR7151490_2.fastq out1=SRR7151490_1_t.fastq out2=SRR71514902_t.fastq ref=/bbmap/resources/adapters.fa ktrim=r k=23 mink=11 hdist=1 tpe tbo
java -ea -Xmx58304m -Xms58304m -cp /bbmap/current/ jgi.BBDuk in1=SRR7151490_1.fastq in2=SRR7151490_2.fastq out1=SRR7151490_1_t.fastq out2=SRR71514902_t.fastq ref=/bbmap/resources/adapters.fa ktrim=r k=23 mink=11 hdist=1 tpe tbo
Executing jgi.BBDuk [in1=SRR7151490_1.fastq, in2=SRR7151490_2.fastq, out1=SRR7151490_1_t.fastq, out2=SRR71514902_t.fastq, ref=/bbmap/resources/adapters.fa, ktrim=r, k=23, mink=11, hdist=1, tpe, tbo]
Version 38.69
maskMiddle was disabled because useShortKmers=true
0.022 seconds.
Initial:
Memory: max=61136m, total=61136m, free=61102m, used=34m
Added 217135 kmers; time: 0.064 seconds.
Memory: max=61136m, total=61136m, free=61027m, used=109m
Input is being processed as paired
Started output streams: 0.027 seconds.
Processing time: 22.848 seconds.
Input: 28291796 reads 2857471396 bases.
KTrimmed: 66418 reads (0.23%) 2072372 bases (0.07%)
Trimmed by overlap: 7444 reads (0.03%) 81230 bases (0.00%)
Total Removed: 12460 reads (0.04%) 2153602 bases (0.08%)
Result: 28279336 reads (99.96%) 2855317794 bases (99.92%)
Time: 22.942 seconds.
Reads Processed: 28291k 1233.19k reads/sec
Bases Processed: 2857m 124.55m bases/sec
real 0m25.604s
user 3m38.195s
sys 0m8.344s
$ singularity run BBTools-38.69.sif bbduk.sh in1=SRR7151490_1_t.fastq in2=SRR71514902_t.fastq qtrim=rl trimq=10 out1=SRR7151490_1_t_qc.fastq out2=SRR7151490_2_t_qc.fastq
java -ea -Xmx58304m -Xms58304m -cp /bbmap/current/ jgi.BBDuk in1=SRR7151490_1_t.fastq in2=SRR71514902_t.fastq qtrim=rl trimq=10 out1=SRR7151490_1_t_qc.fastq out2=SRR7151490_2_t_qc.fastq
Executing jgi.BBDuk [in1=SRR7151490_1_t.fastq, in2=SRR71514902_t.fastq, qtrim=rl, trimq=10, out1=SRR7151490_1_t_qc.fastq, out2=SRR7151490_2_t_qc.fastq]
Version 38.69
0.024 seconds.
Initial:
Memory: max=61136m, total=61136m, free=61102m, used=34m
Input is being processed as paired
Started output streams: 0.021 seconds.
Processing time: 7.409 seconds.
Input: 28279336 reads 2855317794 bases.
QTrimmed: 4386431 reads (15.51%) 183780487 bases (6.44%)
Total Removed: 837690 reads (2.96%) 183780487 bases (6.44%)
Result: 27441646 reads (97.04%) 2671537307 bases (93.56%)
Time: 7.431 seconds.
Reads Processed: 28279k 3805.70k reads/sec
Bases Processed: 2855m 384.26m bases/sec
real 0m8.023s
user 0m39.299s
sys 0m7.407s
BBDuk completed in 25+8 seconds, Trimmomatic in 11 minutes. Certainly a performance difference. How does the quality check out?
Read Quality Control (Visualizing)
Here, we’ve already loaded singularity (above) and moved to the project directory. In this example, I’m going to run FastQC on all of the input files (2), the results from Trimmomatic (4) and the adapter trimmed (2) and quality filtered (2) read pairs of BBDuk.
$ singularity run FastQC-0.11.8.sif SRR7151490_1.fastq SRR7151490_2.fastq SRR7151490_1_t_paired.fastq SRR7151490_1_t_unpaired.fastq SRR7151490_2_t_paired.fastq SRR7151490_2_t_unpaired.fastq SRR7151490_1_t.fastq SRR7151490_1_t_qc.fastq SRR7151490_2_t.fastq SRR7151490_2_t_qc.fastq
Started analysis of SRR7151490_1.fastq
Approx 5% complete for SRR7151490_1.fastq
Approx 10% complete for SRR7151490_1.fastq
Approx 15% complete for SRR7151490_1.fastq
…
…
Approx 95% complete for SRR7151490_1.fastq
Analysis complete for SRR7151490_1.fastq
I’ve omitted the lengthy 5% increments for all 10 files. Basically, FastQC will process each file individually and deposit the results in <filename>_fastqc.zip and <filename>_fastqc.html.
Next, we’ll want to visually summarize these results using MultiQC. I’m running MultiQC in the directory with all the FastQC results, so I’m using “.” to specify “the current directory” on the command line.
$ singularity run MultiQC-1.7.sif .
[INFO ] multiqc : This is MultiQC v1.7
[INFO ] multiqc : Template : default
[INFO ] multiqc : Searching '.'
[INFO ] fastqc : Found 10 reports
[INFO ] multiqc : Compressing plot data
[INFO ] multiqc : Report : multiqc_report.html
[INFO ] multiqc : Data : multiqc_data
[INFO ] multiqc : MultiQC complete
Once that’s done, the resulting directory should look like:
$ ls -lh
# Original input data
SRR7151490_1_fastqc.zip
SRR7151490_2_fastqc.zip
# Trimmomatic results. Paired reads surviving (2) + unpaired reads (mate pair didn't make it) surviving (2)
SRR7151490_1_t_paired_fastqc.zip
SRR7151490_1_t_unpaired_fastqc.zip
SRR7151490_2_t_paired_fastqc.zip
SRR7151490_2_t_unpaired_fastqc.zip
# BBDuk adapter trimming results. BBDuk will only return paired reads with the parameters we specified
SRR7151490_1_t_fastqc.zip
SRR7151490_2_t_fastqc.zip
# BBDuk quality trimming results, using the trimming results (above) as input
SRR7151490_1_t_qc_fastqc.zip
SRR7151490_2_t_qc_fastqc.zip
# MultiQC report and data
multiqc_report.html
multiqc_data
I’ve added comments to the command (above). Normally, this would NOT be in the output, but I’m commenting here to break down what files came from where.
Let’s look at the results here.
Assembly
Assembly isn’t for the faint of heart. It can be frustrating and it can fail for a lot of reasons, often due to insufficient memory or due to the dataset complexity. There’s only so much you can do.
However, our example dataset will finish on OSC within a few hours. Below is the bash script that can be submitted to OSC that should assemble your data.
This will run SPAdes on the Trimmomatic-cleaned reads, alternatives are below.
#SBATCH --time=12:00:00
#SBATCH --nodes=1
#SBATCH --ntasks=40
#SBATCH --job-name=SPAdes_SRR7151490_Trimmomatic
#SBATCH --account=PAS1573
#SBATCH --mail-type=START
#SBATCH --mail-type=END
# Load modules that we'll need
module load singularity
# Set variables for this script - it makes it easier to refer to later on
# Some get confused by this, but in the long run it'll save a lot of typing
# Root/core directories
projectDir="/fs/ess/PAS1573/week7_processing/"
readsDir="${projectDir}/trimmed_trimmomatic/"
spadesLoc="/users/PAS1117/osu9664/eMicro-Apps/SPAdes-3.13.0.sif"
outputDir="${projectDir}/assemblies/SPAdes_with_Trimmomatic"
# Whare are the reads we'll need?
forReads="${readsDir}/SRR7151490_1_t_paired.fastq.gz"
revReads="${readsDir}/SRR7151490_2_t_paired.fastq.gz"
# Assembling with SPAdes, so setting up parameters
genOpts="--meta -k 21,33,55,77" # Paired end, 1 pair only
runOpts="-t 40 -m 190" # Match to job request, 40 cores and ~192 GB of memory
# Now that we have our parameters and input files, we can put everything together
spadesCmd="singularity run ${spadesLoc} ${genOpts} ${runOpts}"
spadesCmd="${spadesCmd} --pe1-1 ${forReads} --pe1-2 ${revReads}"
# I always like to know what command was actually sent to SPAdes
# -o will send the output of SPAdes to the assembly directory, defined above
echo "${spadesCmd} -o ${outputDir}"
${spadesCmd} -o ${outputDir}
Submit using:
sbatch spades_assembly_trimmomatic_reads.sh
Please see the OSC guide for how this job script was created. Since I’m familiar with the sample background (sample complexity, microbes, relative sequencing depth) and the SPAdes assembler for this sample, I can guess at how long to request for the job. I requested 12 hours and a full node (ppn=40 cores, and set SPAdes -t 40). At the end of the run, OSC will let you know what kind of resources you actually used. For this job:
resources_used.vmem=41783760kb
resources_used.walltime=10:37:21
The job required ~42 GB and ~10.5 hours. We can’t do anything about the GB requested, as asking for 40 cores will give you the whole (192 GB) node. We could request 50 GB of memory, but OSC will still charge you for the whole node.
But what if we didn’t want to use Trimmomatic? Let’s use the BBDuk-cleaned reads, generated earlier? Instead of copying-and-pasting the whole bash script, i’ll only highlight the changes:
readsDir=”${projectDir}/trimmed_trimmomatic/” –> readsDir=”${projectDir}/trimmed_bbduk/”
outputDir=”${projectDir}/assemblies/SPAdes_with_Trimmomatic” –> outputDir=”${projectDir}/assemblies/SPAdes_with_BBDuk”
forReads=”${readsDir}/SRR7151490_1_t_paired.fastq.gz” –> forReads=”${readsDir}/SRR7151490_1_t_qc.fastq”
revReads=”${readsDir}/SRR7151490_2_t_paired.fastq.gz” –> revReads=”${readsDir}/SRR7151490_2_t_qc.fastq”
And that’s it! Submit that to OSC!
What resources were used?
resources_used.vmem=42275192kb
resources_used.walltime=10:28:35
About the same amount of resources. It makes sense since our reads were trimmed a tiny amount and there wasn’t much difference between them anyway.
And now, what if we wanted to use a different assembler, let’s say MEGAHIT?
#SBATCH --time=2:00:00
#SBATCH --nodes=1
#SBATCH --ntasks=40
#SBATCH --job-name=METAHIT_SRR7151490_Trimmomatic
#SBATCH --account=PAS1573
#SBATCH --mail-type=START
#SBATCH --mail-type=END
# Load modules that we'll need
module load singularity
# Root/core directories
projectDir="/fs/ess/PAS1573/week7_processing/"
readsDir="${projectDir}/trimmed_trimmomatic/"
megahitLoc="/users/PAS1117/osu9664/eMicro-Apps/MEGAHIT-1.2.8.sif"
outputDir="${projectDir}/assemblies/MEGAHIT_with_Trimmomatic"
# Whare are the reads we'll need?
forReads="${readsDir}/SRR7151490_1_t_paired.fastq.gz"
revReads="${readsDir}/SRR7151490_2_t_paired.fastq.gz"
# Assembling with MEGAHIT, so setting up parameters
genOpts="--k-list 21,41,61,81,99" # K-mer selection is a PhD itself...
runOpts="-t 40 -m 0.9" # Match to job request, 40 cores and 90% of memory
# Now that we have our parameters and input files, we can put everything together
megahitCmd="singularity run ${megahitLoc} ${genOpts} ${runOpts}"
megahitCmd="${megahitCmd} -1 ${forReads} -2 ${revReads}"
# I always like to know what command was actually sent to SPAdes
# -o will send the output of SPAdes to the assembly directory, defined above
echo "${megahitCmd} -o ${outputDir}"
${megahitCmd} -o ${outputDir}
Notice how I only changed a few lines, as when we switched from using Trimmomatic to BBDuk cleaned reads. The lines that were changed:
megahitLoc=”/users/PAS1117/osu9664/eMicro-Apps/MEGAHIT-1.2.8.sif”
outputDir=”${projectDir}/assemblies/MEGAHIT_with_Trimmomatic”
genOpts=”–k-list 21,41,61,81,99”
runOpts=”-t 40 -m 0.9” # Match to job request, 40 cores and 90% of memory
megahitCmd=”singularity run ${megahitLoc} ${genOpts} ${runOpts}”
megahitCmd=”${megahitCmd} -1 ${forReads} -2 ${revReads}”
The location of the assembler changed, as did the output directory. Also, because we’re using a different assembler with different parameters/arguments, we need to change those as well.
Submit!
resources_used.vmem=7302672kb
resources_used.walltime=00:20:29
Wow. 20 minutes and 7 GB of memory. More on this later…
And finally, IDBA-UD. Oh wait, IDBA-UD wants fasta-formatted sequences. We need to make a small change.
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/IDBA-UD-1.1.3.sif fq2fa --merge --filter SRR7151490_1_t_paired.fastq SRR7151490_2_t_paired.fastq SRR7151490_t_paired.fasta
real 1m9.317s
user 1m3.504s
sys 0m4.836s
We just converted our gzip-decompressed fastq files into fasta. Now we can submit. Let’s use BBDuk reads this time.
#SBATCH --time=2:00:00
#SBATCH --nodes=1
#SBATCH --ntasks=40
#SBATCH --job-name=IDBA_UD_SRR7151490_BBDuk
#SBATCH --account=PAS1573
#SBATCH --mail-type=START
#SBATCH --mail-type=END
# Load modules that we'll need
module load singularity
# Root/core directories
projectDir="/fs/ess/PAS1573/week7_processing/"
readsDir="${projectDir}/trimmed_bbduk/"
idbaLoc="/users/PAS1117/osu9664/eMicro-Apps/IDBA-UD-1.1.3.sif"
outputDir="${projectDir}/assemblies/IDBA_UD_with_BBDuk"
# Whare are the reads we'll need?
bothReads="${readsDir}/SRR7151490_t_paired.fasta"
# Assembling with IDBA-UD, so setting up parameters
runOpts="--num_threads 40"
idbaCmd="singularity run ${idbaLoc} ${runOpts}"
idbaCmd="${idbaCmd} -r ${bothReads}"
echo "${idbaCmd} -o ${outputDir}"
${idbaCmd} -o ${outputDir}
And resources used:
resources_used.vmem=56120080kb
resources_used.walltime=01:29:35
Binning
For all the binning tools, we’re going to use the SPAdes assembly results that used the Trimmomatic-cleaned reads. Why? Because it’s the one I selected - no rhyme or reason. Feel free to choose SPAdes w/ BBDuk, or Megahit with BBDuk, or whichever assembler+QC reads you want.
The first thing we need to do is run read mapping. We’ll be mapping reads from our QC pipeline against our assembled contigs. We do this because all the binning tools [we’ll be discussing] require read coverage information, generated from read mapping.
We’ll use bowtie2 to do our read mapping. Alternatively, we could use [an equally good] BWA, and I selected bowtie2 just because we use it more in the lab. The first step is to create a bowtie2-indexed database file using our contigs from SPAdes.
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/BamM-1.7.0.sif bowtie2-build -f /fs/ess/PAS1573/week7_processing/assemblies/SPAdes_with_Trimmomatic/contigs.fasta contigs_bowtie2
Next, we map our reads using bowtie2.
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/BamM-1.7.0.sif bowtie2 -q --phred33 --end-to-end -p 40 -I 0 -X 2000 --no-unal -x contigs_bowtie2 -1 /fs/ess/PAS1573/week7_processing/trimmed_trimmomatic/SRR7151490_1_t_paired.fastq.gz -2 /fs/ess/PAS1573/week7_processing/trimmed_trimmomatic/SRR7151490_2_t_paired.fastq.gz -S SPades_contigs_vs_SRR7151490.sam
Convert the sam file into a bam via samtools. We’re essentially “viewing” with an input format of SAM and outputting a BAM format. Notice the use of the “>” redirect.
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/BamM-1.7.0.sif samtools view -Sb SPades_contigs_vs_SRR7151490.sam > SPades_contigs_vs_SRR7151490.bam
Now, sort… because most/many softwares require sorted BAM files.
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/BamM-1.7.0.sif samtools sort -o SPades_contigs_vs_SRR7151490.sorted.bam SPades_contigs_vs_SRR7151490.bam
Now we can run our 1st tool, MetaBAT2. We’ll use a convenient wrapper script, provided with MetaBAT2, that will run the entire Metabat pipeline.
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/MetaBAT2-2.14.sif runMetaBat.sh /fs/ess/PAS1573/week7_processing/assemblies/SPAdes_with_Trimmomatic/contigs.fasta SPades_contigs_vs_SRR7151490.sorted.bam
Executing: 'jgi_summarize_bam_contig_depths --outputDepth contigs.fasta.depth.txt --percentIdentity 97 --minContigLength 1000 --minContigDepth 1.0 --referenceFasta /fs/ess/PAS1573/week7_processing/assemblies/SPAdes_with_Trimmomatic/contigs.fasta SPades_contigs_vs_SRR7151490.sorted.bam' at Mon Oct 7 16:26:18 UTC 2019
Output depth matrix to contigs.fasta.depth.txt
Minimum percent identity for a mapped read: 0.97
minContigLength: 1000
minContigDepth: 1
Reference fasta file /fs/ess/PAS1573/week7_processing/assemblies/SPAdes_with_Trimmomatic/contigs.fasta
jgi_summarize_bam_contig_depths v2.14 2019-10-01T04:25:59
Output matrix to contigs.fasta.depth.txt
Reading reference fasta file: /fs/ess/PAS1573/week7_processing/assemblies/SPAdes_with_Trimmomatic/contigs.fasta
... 484282 sequences
0: Opening bam: SPades_contigs_vs_SRR7151490.sorted.bam
Processing bam files
Thread 0 finished: SPades_contigs_vs_SRR7151490.sorted.bam with 8146462 reads and 7177777 readsWellMapped
Creating depth matrix file: contigs.fasta.depth.txt
Closing most bam files
Closing last bam file
Finished
Finished jgi_summarize_bam_contig_depths at Mon Oct 7 16:28:49 UTC 2019
Creating depth file for metabat at Mon Oct 7 16:28:49 UTC 2019
Executing: 'metabat2 --inFile /fs/ess/PAS1573/week7_processing/assemblies/SPAdes_with_Trimmomatic/contigs.fasta --outFile contigs.fasta.metabat-bins-20191007_162849/bin --abdFile contigs.fasta.depth.txt' at Mon Oct 7 16:28:49 UTC 2019
MetaBAT 2 (v2.14) using minContig 2500, minCV 1.0, minCVSum 1.0, maxP 95%, minS 60, and maxEdges 200.
8 bins (22664127 bases in total) formed.
The next tool we’re going to use is MaxBin2. It’s probably the easiest of the 3 binners we’ll use as it can handle the read mapping component in the background. Of course, we could re-use the bam files, generated earlier for MetaBAT2, but for this example we’ll be letting MaxBin2 handle everything!
$ singularity run /users/PAS1117/osu9664/eMicro-Apps/MaxBin2-2.2.6.sif -contig /fs/ess/PAS1573/week7_processing/assemblies/SPAdes_with_Trimmomatic/contigs.fasta -reads /fs/ess/PAS1573/week7_processing/trimmed_trimmomatic/SRR7151490_1_t_paired.fastq.gz -reads2 /fs/ess/PAS1573/week7_processing/trimmed_trimmomatic/SRR7151490_2_t_paired.fastq.gz -out maxbin2
The last binning tool we’ll use is CONCOCT. Before getting started, we’re going to index our BAM-sorted file, so CONCOCT can (later) generate a coverage table from it. For this, we’ll continue to use the BamM singularity container because it has samtools already. In this example, I’ve also copied the BAM file from its earlier location (see setup, above) to our current location.
$ cp ../spades_trimmomatic_metabat2/SPades_contigs_vs_SRR7151490.sorted.bam .
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/BamM-1.7.0.sif samtools index SPades_contigs_vs_SRR7151490.sorted.bam
Now we need to cut up our contigs into 10-kb chunks.
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/CONCOCT-1.1.0.sif cut_up_fasta.py /fs/ess/PAS1573/week7_processing/assemblies/SPAdes_with_Trimmomatic/contigs.fasta -c 10000 -o 0 --merge_last -b contigs_10K.bed > contigs_10K.fa
Next, we need to generate a CONCOCT-formatted coverage table from our BAM file.
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/CONCOCT-1.1.0.sif concoct_coverage_table.py contigs_10K.bed SPades_contigs_vs_SRR7151490.sorted.bam > coverage_table.tsv
Now we can finally run CONCOCT!
$ singularity run /users/PAS1117/osu9664/eMicro-Apps/CONCOCT-1.1.0.sif --threads 40 --composition_file contigs_10K.fa --coverage_file coverage_table.tsv -b concoct_output/
Up and running. Check /fs/ess/PAS1573/week7_processing/bins/spades_trimmomatic_concoct/concoct_output/log.txt for progress
29628 926 40
Setting 40 OMP threads
Generate input data
0,-2157228.566412,77771.939575
1,-2108159.159213,49069.407199
2,-2061582.303224,46576.855989
3,-1994050.599227,67531.703998
4,-1933958.933656,60091.665571
5,-1851248.853164,82710.080491
6,-1745647.721013,105601.132151
7,-1648890.787478,96756.933535
...
...
495,-1302524.814091,0.337423
496,-1302525.131015,0.316925
497,-1302524.727849,0.403166
498,-1302515.943374,8.784475
499,-1302497.827711,18.115663
Note: I have trimmed the output from the program. Assume that there are lines for 8 - 494, above. Almost the last step, and for now, we need to merge the clustering results (as we split it up earlier).
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/CONCOCT-1.1.0.sif merge_cutup_clustering.py concoct_output/clustering_gt1000.csv > concoct_output/clustering_merged.csv
No consensus cluster for contig NODE_13_length_87447_cov_2.121758: [('10', 1), ('35', 7)] Chosen cluster: 35
real 0m39.468s
user 0m0.148s
sys 0m0.185s
Looks like CONCOCT couldn’t decide on where to place a contig. Sometimes this will happen - the binning tool isn’t perfect (well, none of them are) and it’s just what you get with environmental data.
Finally, we need to extract each of the bins as fasta files (this makes downstream work easier).
$ singularity exec /users/PAS1117/osu9664/eMicro-Apps/CONCOCT-1.1.0.sif extract_fasta_bins.py /fs/ess/PAS1573/week7_processing/assemblies/SPAdes_with_Trimmomatic/contigs.fasta concoct_output/clustering_merged.csv --output_path concoct_output/fasta_bins
real 0m14.276s
user 0m7.543s
sys 0m1.313s
And now that we finally have our bins, let’s check the quality with CheckM. For this we’re only going to use the MetaBat2 bins, but you can use any individual binning tool or the aggregated/consolidated results from DAS_Tool or MetaWRAP (that we’ll do below).
$ singularity run /users/PAS1117/osu9664/eMicro-Apps/CheckM-1.0.18.sif lineage_wf -t 40 -x fa bins/spades_trimmomatic_metabat2/contigs.fasta.metabat-bins-20191007_162849 checkm_spades_trimmomatic_metabat2
[2019-10-08 04:29:16] INFO: CheckM v1.0.18
[2019-10-08 04:29:16] INFO: checkm lineage_wf -t 40 -x fa bins/spades_trimmomatic_metabat2/contigs.fasta.metabat-bins-20191007_162849 checkm_spades_trimmomatic_metabat2
[2019-10-08 04:29:16] INFO: [CheckM - tree] Placing bins in reference genome tree.
[2019-10-08 04:29:17] INFO: Identifying marker genes in 8 bins with 40 threads:
Finished processing 8 of 8 (100.00%) bins.
[2019-10-08 04:29:37] INFO: Saving HMM info to file.
[2019-10-08 04:29:37] INFO: Calculating genome statistics for 8 bins with 40 threads:
Finished processing 8 of 8 (100.00%) bins.
[2019-10-08 04:29:37] INFO: Extracting marker genes to align.
[2019-10-08 04:29:37] INFO: Parsing HMM hits to marker genes:
Finished parsing hits for 8 of 8 (100.00%) bins.
[2019-10-08 04:29:38] INFO: Extracting 43 HMMs with 40 threads:
Finished extracting 43 of 43 (100.00%) HMMs.
[2019-10-08 04:29:38] INFO: Aligning 43 marker genes with 40 threads:
Finished aligning 43 of 43 (100.00%) marker genes.
[2019-10-08 04:29:39] INFO: Reading marker alignment files.
[2019-10-08 04:29:39] INFO: Concatenating alignments.
[2019-10-08 04:29:39] INFO: Placing 8 bins into the genome tree with pplacer (be patient).
[2019-10-08 04:33:37] INFO: { Current stage: 0:04:21.041 || Total: 0:04:21.041 }
[2019-10-08 04:33:37] INFO: [CheckM - lineage_set] Inferring lineage-specific marker sets.
[2019-10-08 04:33:37] INFO: Reading HMM info from file.
[2019-10-08 04:33:37] INFO: Parsing HMM hits to marker genes:
Finished parsing hits for 8 of 8 (100.00%) bins.
[2019-10-08 04:33:38] INFO: Determining marker sets for each genome bin.
Finished processing 8 of 8 (100.00%) bins (current: bin.2).
[2019-10-08 04:33:39] INFO: Marker set written to: checkm_spades_trimmomatic_metabat2/lineage.ms
[2019-10-08 04:33:39] INFO: { Current stage: 0:00:01.964 || Total: 0:04:23.006 }
[2019-10-08 04:33:39] INFO: [CheckM - analyze] Identifying marker genes in bins.
[2019-10-08 04:33:40] INFO: Identifying marker genes in 8 bins with 40 threads:
Finished processing 8 of 8 (100.00%) bins.
[2019-10-08 04:34:46] INFO: Saving HMM info to file.
[2019-10-08 04:34:46] INFO: { Current stage: 0:01:06.748 || Total: 0:05:29.754 }
[2019-10-08 04:34:46] INFO: Parsing HMM hits to marker genes:
Finished parsing hits for 8 of 8 (100.00%) bins.
[2019-10-08 04:34:47] INFO: Aligning marker genes with multiple hits in a single bin:
Finished processing 8 of 8 (100.00%) bins.
[2019-10-08 04:34:52] INFO: { Current stage: 0:00:06.238 || Total: 0:05:35.993 }
[2019-10-08 04:34:52] INFO: Calculating genome statistics for 8 bins with 40 threads:
Finished processing 8 of 8 (100.00%) bins.
[2019-10-08 04:34:53] INFO: { Current stage: 0:00:00.299 || Total: 0:05:36.292 }
[2019-10-08 04:34:53] INFO: [CheckM - qa] Tabulating genome statistics.
[2019-10-08 04:34:53] INFO: Calculating AAI between multi-copy marker genes.
[2019-10-08 04:34:53] INFO: Reading HMM info from file.
[2019-10-08 04:34:54] INFO: Parsing HMM hits to marker genes:
Finished parsing hits for 8 of 8 (100.00%) bins.
--------------------------------------------------------------------------------------------------------------------------------------------------------------------
Bin Id Marker lineage # genomes # markers # marker sets 0 1 2 3 4 5+ Completeness Contamination Strain heterogeneity
--------------------------------------------------------------------------------------------------------------------------------------------------------------------
bin.8 k__Bacteria (UID203) 5449 104 58 2 51 51 0 0 0 97.41 43.09 9.80
bin.4 p__Actinobacteria (UID1454) 732 200 117 8 191 1 0 0 0 93.59 0.43 100.00
bin.6 k__Bacteria (UID3187) 2258 188 117 85 84 15 4 0 0 59.36 16.20 7.41
bin.2 k__Bacteria (UID203) 5449 104 58 65 34 5 0 0 0 51.90 6.38 20.00
bin.3 k__Bacteria (UID2982) 88 227 146 131 93 3 0 0 0 46.53 2.05 0.00
bin.1 k__Bacteria (UID203) 5449 104 58 79 23 2 0 0 0 32.04 2.07 0.00
bin.5 k__Bacteria (UID203) 5449 104 58 88 16 0 0 0 0 15.60 0.00 0.00
bin.7 k__Bacteria (UID203) 5449 104 58 94 10 0 0 0 0 15.52 0.00 0.00
--------------------------------------------------------------------------------------------------------------------------------------------------------------------
[2019-10-08 04:34:55] INFO: { Current stage: 0:00:02.063 || Total: 0:05:38.356 }
real 5m39.476s
user 8m47.760s
sys 0m44.529s
As you can see, of the 8 bins determined by MetaBAT2, a few of them are more-or-less complete. One of the more complete genomes has a bit of contamination. Most of the other bins don’t have nearly as much contamination, but also aren’t nearly as complete. With a single metagenome and just testing these tools with real data, that’s pretty good. However, there’s a lot of things we can do with these results - not just post-binning analysis, but also re-assembly with different parameters or using bin refinement tools, such as MetaWRAP and DAS_Tool to get the best bins from the three different binning tools. This aggregation often results in superior bins - better than any binner can do individually.
For the purposes of this exercise, we’ll be using DAS_Tool instead of MetaWRAP. For members of the Sullivan lab, MetaWRAP is installed to the lab project directory via modules. For everyone else (on OSC, or eMicro), DAS_Tool is available and instructions follow!
Bin Refinement
We’ll use DAS Tool to refine our bins generated from CONCOCT, MetaBAT2 and MaxBin2. Half the work is formatting the input files, so we’ll start with that.
Convert the CONCOCT csv into a tsv.
$ perl -pe "s/,/\tconcoct./g;" spades_trimmomatic_concoct/concoct_output/clustering_merged.csv > spades_trimmomatic_das_tool/concoct.scaffolds2bin.tsv
Note: Keep in mind that we’re using clustering_merged.csv.
Now to convert MetaBAT2 into a tsv.
$ /users/PAS1117/osu9664/eMicro-Apps/Fasta_to_Scaffolds2Bin.sh -i spades_trimmomatic_metabat2/contigs.fasta.metabat-bins-20191007_162849 -e fa > spades_trimmomatic_das_tool/metabat2.scaffolds2bin.tsv
And now MaxBin2…
$ /users/PAS1117/osu9664/eMicro-Apps/Fasta_to_Scaffolds2Bin.sh -i spades_trimmomatic_maxbin2 -e fasta > spades_trimmomatic_das_tool/maxbin.scaffolds2bin.tsv
Finally, let’s run DAS Tool!
$ singularity run /users/PAS1117/osu9664/eMicro-Apps/DAS_Tool-.sif -i spades_trimmomatic_das_tool/concoct.scaffolds2bin.tsv,spades_trimmomatic_das_tool/maxbin.scaffolds2bin.tsv,spades_trimmomatic_das_tool/metabat2.scaffolds2bin.tsv -l concoct,maxbin,metabat -c /fs/ess/PAS1573/week7_processing/assemblies/SPAdes_with_Trimmomatic/contigs.fasta -o spades_trimmomatic_das_tool/DAS_Tool_results --search_engine diamond --threads 40
/miniconda3/bin//DAS_Tool: line 241: usearch: command not found
Running DAS Tool using 40 threads.
predicting genes using Prodigal V2.6.3: February, 2016
identifying single copy genes using diamond version 0.9.14
During startup - Warning messages:
1: Setting LC_CTYPE failed, using "C"
2: Setting LC_COLLATE failed, using "C"
3: Setting LC_TIME failed, using "C"
4: Setting LC_MESSAGES failed, using "C"
5: Setting LC_MONETARY failed, using "C"
6: Setting LC_PAPER failed, using "C"
7: Setting LC_MEASUREMENT failed, using "C"
WARNING: Some scaffold names ofspades_trimmomatic_das_tool/concoct.scaffolds2bin.tsv do not match assembly headers:
contig_id
evaluating bin-sets
starting bin selection from 39 bins
|||
bin selection complete: 3 bins above score threshold selected.
real 0m56.315s
user 17m52.350s
sys 0m15.872s
So there were only 3 bins that exceeded our [default] thresholds. We can work with that. What are the 3 bins?
And what are the results?
$ ls -lh spades_trimmomatic_das_tool/
total 165M
maxbin.scaffolds2bin.tsv
metabat2.scaffolds2bin.tsv
concoct.scaffolds2bin.tsv
DAS_Tool_results_proteins.faa
DAS_Tool_results_proteins.faa.bacteria.scg
DAS_Tool_results_proteins.faa.archaea.scg
DAS_Tool_results.seqlength
DAS_Tool_results_concoct.eval
DAS_Tool_results_maxbin.eval
DAS_Tool_results_metabat.eval
DAS_Tool_results_DASTool_scaffolds2bin.txt
DAS_Tool_results_DASTool_summary.txt
DAS_Tool_results_DASTool.log
DAS_Tool_results_DASTool_hqBins.pdf
DAS_Tool_results_DASTool_scores.pdf
Now we’ve gone from raw sequence data, downloaded from NCBI SRA to bin-refined MAGs that have been quality-checked with CheckM. Only a little further and the manuscript will be published!